Genomics Pipelines, Tools, and Hackathons
A list of my genomics pipelines and tools developed at SCRI, Fred Hutch, and through external collaborations, such as Hackathons. These pipelines and tools were used to reproducibly process data or generate figures for many of the published journal articles on projects I supported.
Genomics Data Pipelines
1. Cut and Run Nextflow workflow
- Custom pipeline developed from nf-core workflow template and uses most nf-core best practices.
Show Details
Performs Bowtie2 alignment, quality trimming of reads with trimgalore, SEACR peak calling, and optionally MACS2 peak calling.
MACS2 requires an effective genome size to call peaks, which can be provided directly in
nextflow.configor useunique-kmers.pyto calculate the effective genome size on the fly.Coverage tracks are produced for visualization in IGV.
Performs general QC statistics on the fastqs with fastqc, the alignment, peak calling, and sample similarity using deeptools. Finally, the QC reports are collected into a single file using multiQC.
Documented using parameterized Rmd that knits to HTML and github markdown formats.
CI/CD with Atlassian bamboo agent for functional tests using small toy datasets and to re-build documentation.
Public version can be found here, but most recent releases are private repos at SCRI bitbucket.

2. RNA-seq Quantification Nextflow workflow
- Custom RNA-seq alignment and quantification pipeline developed using nf-core tools and uses most nf-core best practices.
Show Details
Designed to output gene expression counts from bulk RNA-seq using
STARaligner using--quantmode.Performs general QC statistics on the fastqs with
fastqcand the alignment usingrseqc.Finally, the QC reports are collected into a single file using
multiQC.
Documented using parameterized Rmd that knits to HTML and github markdown formats.
Public version can be found here, but most recent releases are private repos at SCRI bitbucket.

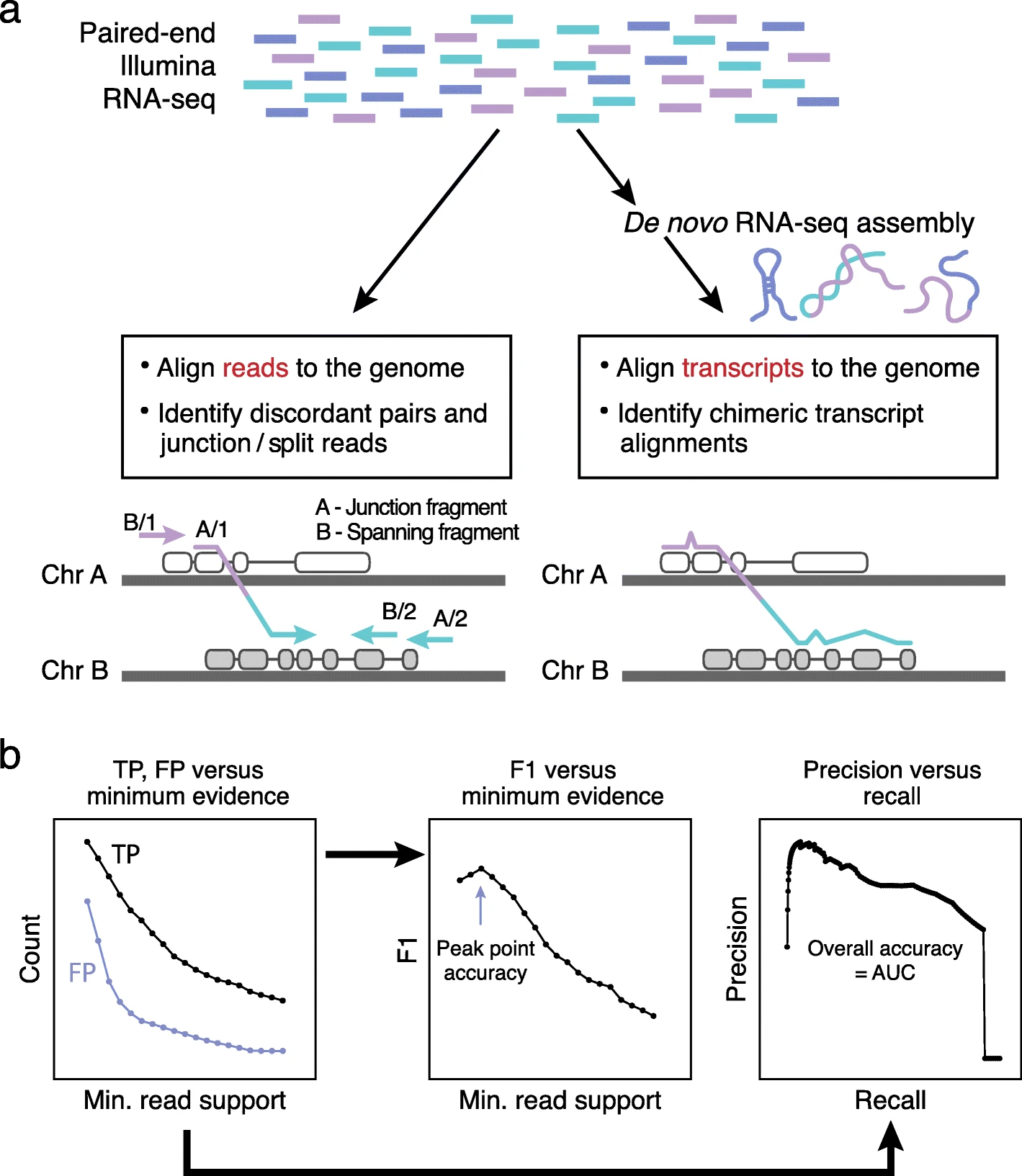
3. RNA-seq Fusion Detection Nextflow workflow
- Custom RNA-seq pipeline to detect fusion transcripts developed using Nextflow DSL2 syntax.
Show Details
Bulk RNA-seq fusion detection pipeline with
STARaligner,STAR-Fusion, andFusion-Inspector. This includes the most relevant output files, such as SJ.out.tab, aligned.bam, and chimeric.junctions.tab, and the fusion inspector HTML report.The workflow also includes the
CICEROfusion detection algorithm that is run using the aligned.bam from STAR-aligner output.In addition, the fastq files undergo quality control checks and a multiQC report.
Configured for SLURM, PBS pro, and AWS batch executors with containerized software. Outputs can be saved locally or configured to upload data to AWS S3 buckets.
Public version can be found here, but most recent releases are private repos at SCRI bitbucket.
Other Pipelines
Other nextflow pipelines generated with nextflow and nf-core tools include:
CutandRun and Chip-seq heatmaps (AKA tornado plots - see Figure below)
VCF variant annotation
splitting ATAC-seq bams by fragment size

CI/CD and Reproducible Development Environments
Use of Github actions and Gitpod with DevContainers to automate tasks, like testing and building of applications or websites, as well as create shareable and reproducible development environments.
Reproducible development environments allow team members to actively contribute to a shared code repository, each each team member using an identical compute environment by enabling the use of a containerized environment with devcontainers.
Github Actions
Personal website found here
- Uses a ‘on push’ trigger with github hosted runners
- the repository is checked out and cloned on the remote the ubuntu build machine (a gh hosted runner)
- install
quartoon the ubuntu build machine - use
quarto renderto build the website - the workflow artifacts (a tar archive of the rendered website files) are uploaded to a temporary server
- deploy the artifacts (rendered website) to the github pages URL
Gitpod Development Environment
I am working on the DevOps for Data Science lab exercises and recording the work in a github repository using
- environment is generated directly from a clone of the public repository
- a containerized environment is then built using Docker, which is orchestrated by GitPod Desktop
- automations are used to install required dependencies:
- a reproducible R environment is managed with Renv package to install R packages
- a reproducible Python environment is managed with venv library to install python packages
- the entire project and identical compute environment can be collaboratively developed using this link
Technical Skills and Courses
Additional details about technical skills, including course completion or course progress:
-
Git and Github
Docker
Linux
Python
SQL
AI and Data Scientist
Data Analyst
-
Postgres SQL Database for Toy Worldcup Data
Postgres SQL Database for Toy Student Data
Postgres SQL Database for Toy Intergalactic Data
Additional course/module completions listed in my profile
-
- SQL programming course
- Introduction to SQL Queries and relational databases
- Completed: Nov 2024
- Introduction to Linux: Users and Permissions
- Useful refresher course
- Completed: Jan 2025
- SQL programming course
Hackathon Projects
2024 NF-Core Nextflow Hackathon
Created institutional profile on nf-core github
Added nf-validation tests to gunzip modules
2021 Bringing Genomics Data to the Clinic Hackathon
2018 Consensus Machine Learning for Gene Target Selection in Pediatric AML Risk: NCBI Hackathon
R Packages
- RNA-seq and multi-omics Data Analysis
R package was developed for use in the Meshinchi lab to help streamline the association of clinical covariates and RNA-seq and miRNA-seq expression data.
- RNA-seq Fusion Breakpoint Data Analysis
R package for Sequence Search in BAM files using R Bioconductor
Bioinformatics / Statistical Analysis Notebooks
The analysis notebooks (primarily Rmarkdown) for all analyses from Fred Hutch can be found at Meshinchi Lab. Selected analysis notebooks and scripts from SCRI are hosted at Research Scientific Computing github repository.
References
These pipelines and tools were used to reproducibly process data for many of the published journal articles on projects I supported. See the publications page for details.